The following case is featured in the "You Make The Diagnosis" section of the December 2024 Optometric Retina Society quarterly e-newsletter, which can be found here.
A 72-year-old white male presented for his bioptic driving recertification. He was an established patient at the low vision clinic with a previous diagnosis of degenerative drusen of the macula, which had been noted for over a decade. The patient did not have any pertinent medical history, but he did report taking Preservision vitamins as recommended by a previous provider. There was scarce imaging performed in prior exams as the focus of the previous visits had been on low vision assessment and device trials. The patient reported having stable vision growing up, or at least he did not feel his vision was particularly poor in childhood. He discovered how poor his vision had become in his fifties when he attempted to renew his driver's license but failed the vision screening test. He reported a brother with similar vision impairment and thought he was also diagnosed with age-related macular degeneration.
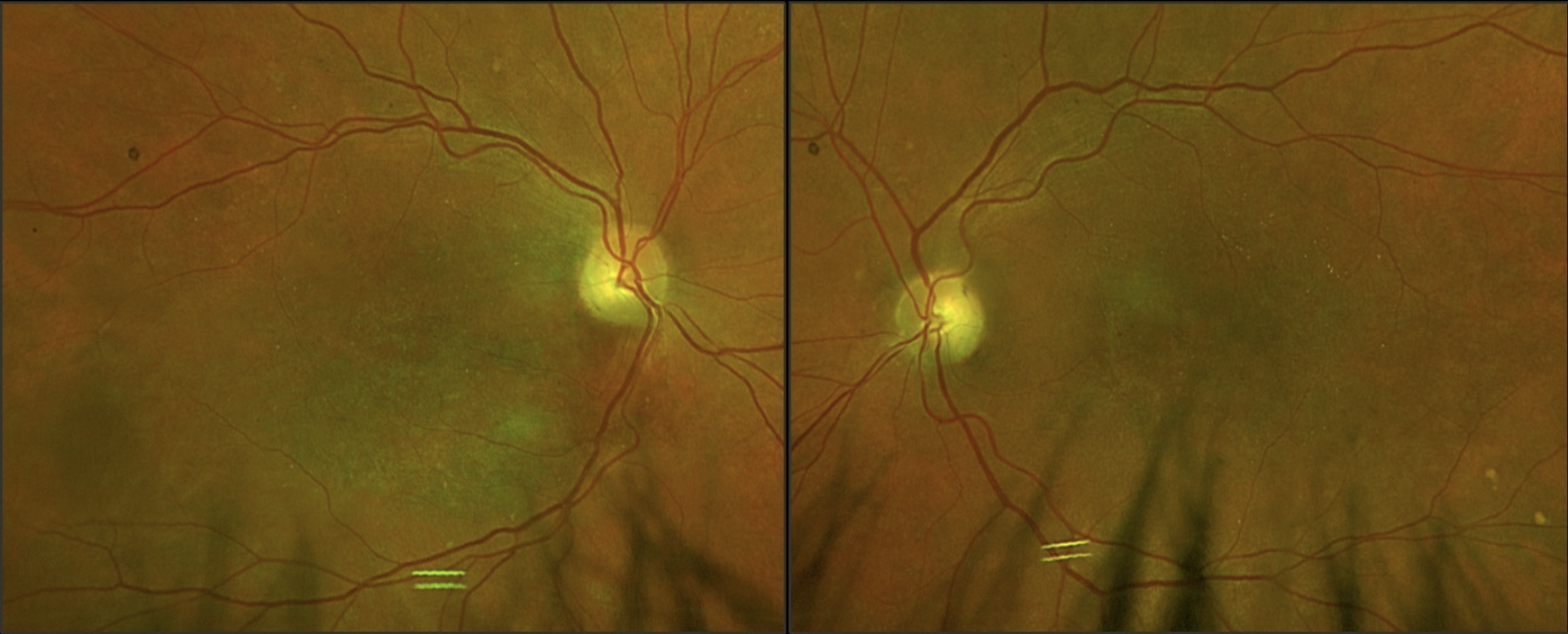 |
| Image 1: Fundus photos of OD (left) and OS (right) acquired on Optos. Note the scattered small hard drusen-like deposits around the arcades and general macular depigmentation with mottling. Click image to enlarge. |
At this visit, vision was comparable to prior testing with acuities of 20/200 OD, 20/150+ OS. With a mounted bioptic telescope, the patient achieved 20/40 OD. Esterman binocular visual field testing revealed a full, clear field of 160 degrees horizontally OU. All other entrance testing and anterior segment findings were unremarkable. Upon examination of the fundus, scattered small drusen were notable throughout the posterior pole and arcades. The maculae had significant pigment mottling with rare drusen (image 1) and an epiretinal membrane in the left eye.
 |
|
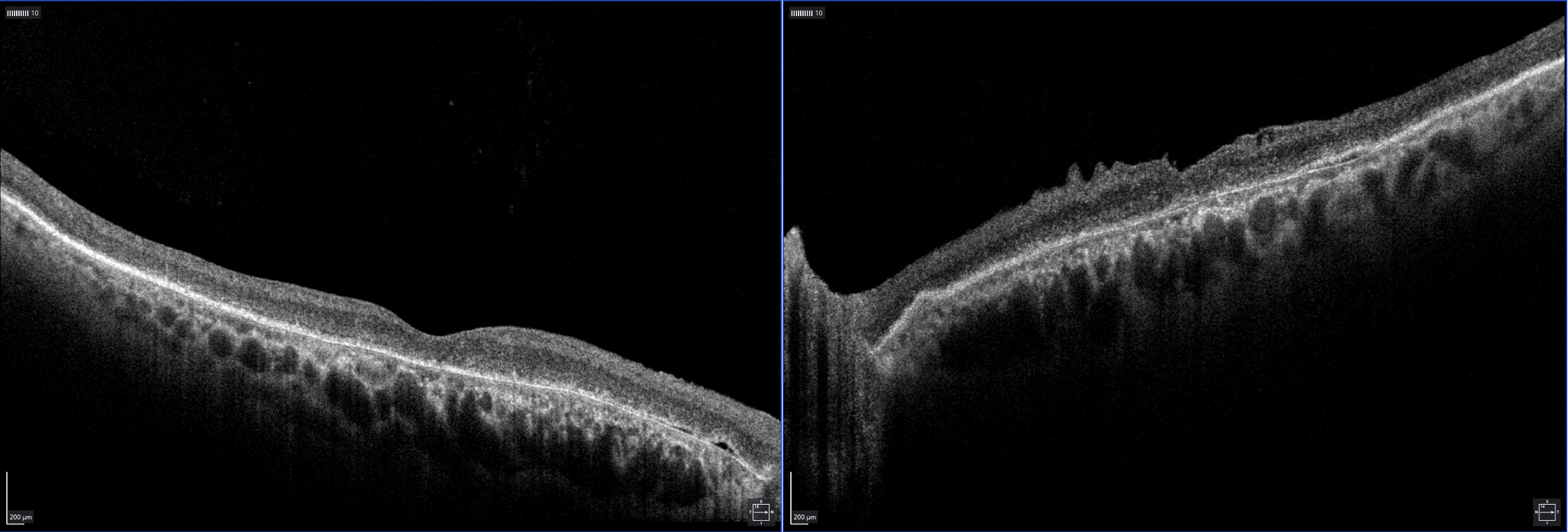
Image 2: OCT of macula OD (left), OS (right) displaying general atrophy of the outer retinal layers and shallow PEDs. An incidental ERM is noted OS. Click image to enlarge. |
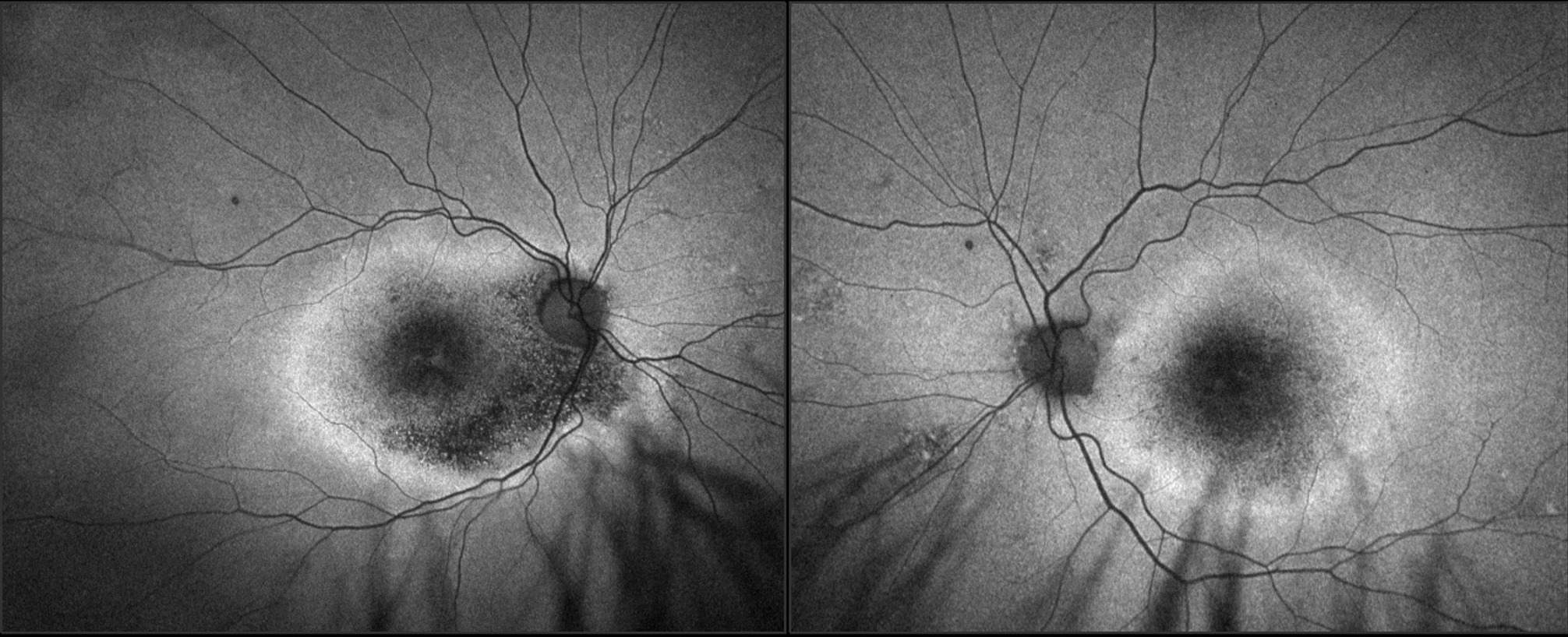
OCT revealed diffuse retinal atrophy, specifically absent or very disrupted outer retinal layers and few scattered shallow PEDs. No significant drusen deposits were noted centrally (image 2). Widefield fundus photos were acquired on Optos with fundus autofluorescence (FAF) and revealed central hypofluorescence with a hyperfluorescent ring surround (image 3). The appearance on OCT and Optos was fairly symmetric between the eyes.
 |
|
Image 3: FAF imaging acquired with Optos of OD, OS. Note significant hypofluorescence OU, with distressed RPE surrounding concentrically noted by hyperfluorescence. The appearance is symmetric between the eyes, with slightly more hypofluorescence OD. Click image to enlarge. |
The clinical findings did not correlate with those of AMD. The patient denied taking any medications associated with retinopathy. The symmetric fundus examination, FAF, and OCT was suspicious for a hereditary retinal dystrophy. At this point in the exam, genetic testing was offered to provide insight into the cause of these retinal findings.
An inherited retinal disorder panel, testing 330 genes, was ordered. The results revealed a significant pathogenic variant in the RS1 gene, which is associated with X-Linked Juvenile Retinoschisis (XLRS).1 RS1 codes for a protein called retinoschisin, that aids cell adhesion and is produced by bipolar and photoreceptor cells. Its particular folding structure can be disrupted by various mutations of exons or other regions within the gene which affect the eight subunits that give the protein its adhesive qualities.2 This patient in particular had a variant in exon 1 of his RS1 gene. Although no significant intraretinal fluid was noted on the exam, this is not entirely unusual for XLRS, especially in later stages of the disorder.
Studies have shown that in later decades of life, patients with XLRS convert from the stereotypical presentation of intraretinal cystic cavities to a general central atrophic appearance over time.3,4 Between the second and third decades of life, atrophy of the RPE and general pigmentary changes/mottling occur, as demonstrated in this case. Visual acuity outcomes may vary depending on the retinal structural changes that occur over time, but typically there is progressive loss of acuity in the earlier decades, stabilization of acuities in the second decade, and then decline in the fifth to sixth decades.5 This correlates to the patient's stated ocular history, in which he only noted significant decline in vision in his fifties, when he failed his driver's license renewal test. The X-linked genetic inheritance pattern also raised concern that his brother could be affected by the same mutation, as males are more prone to exhibit the X-linked mutation.
In this case, genetic counseling was offered to the patient. He did not report having any male offspring nor living female offspring who could have carrier status. Genetic testing and examination were offered to his brother, but they elected not to pursue. Other management included continuing his care in the low vision clinic by recertifying his bioptic driver's license. Additionally, the patient was counseled that an AREDS vitamin would not help his condition, and he could discontinue.
Utilizing genetic testing can be a powerful tool to help correctly identify curiously presenting retinal disorders and in turn avoid misdiagnosis. In this case, the patient was taking expensive vitamin supplements for many years, which ultimately had no impact on the progression of his condition. Additionally, he was unaware of the inheritability of his condition and the likelihood of it affecting other family members. By offering genetic testing, the family can better understand who may be at risk of having the condition and better prepare for those outcomes.

