 |
A 76-year old European female patient presented for evaluation of chronic blurry vision OS for about 12 months. She carries a diagnosis of central retinal vein occlusion status post multiple intravitreal bevacizumab injections. There was previous concern for giant cell arteritis, and she underwent subsequent temporal artery biopsy, which was negative. She previously underwent uncomplicated cataract surgery OU. Systemic history included stage I diffuse large B-cell lymphoma diagnosed two years prior that was treated with chemotherapy and radiation, and the patient has been in remission since completing treatment.
Her presenting visual acuity (VA) was 20/30 OD and 20/200 OS, with no improvement with pinhole OU. Intraocular pressures were 16mm Hg OD and 20mm Hg OS, extraocular motilities were full and symmetric OU and there was a relative afferent pupillary defect OS. Slit lamp exam showed well-positioned posterior chamber intraocular lenses OU and 3+ vitreous cells present in sheets OS.
Take the Retina Quiz
1. What is the most likely diagnosis?
a. Birdshot chorioretinitis.
b. Metastatic uveal lymphoma.
c. Primary uveal lymphoma.
d. Primary vitreoretinal lymphoma.
2. All of the following are typical features of this disease, except:
a. Leopard spotted appearance on fundus auto fluorescence (FAF).
b. Multifocal yellow-white sub-RPE lesions.
c. Posterior synechiae.
d. Vitreous cells.
3. What is the most appropriate next step?
a. Diagnostic pars plana vitrectomy.
b. Enucleation.
c. Observation.
d. Oral prednisone.
4. Which of the following may be suggestive, or confirmatory, of this patient’s disease?
a. CD20-positive B-cells.
b. Elevated interleukin-10 in the aqueous.
c. MYD88 gene mutation.
d. All of the above.
5. Which of the following is true regarding prognosis for this disease?
a. Diagnosis is challenging and may be delayed due to misdiagnosis.
b. Ophthalmic intervention does not impact systemic prognosis.
c. Prognosis is poor with high rates of morbidity and mortality.
d. All of the above.
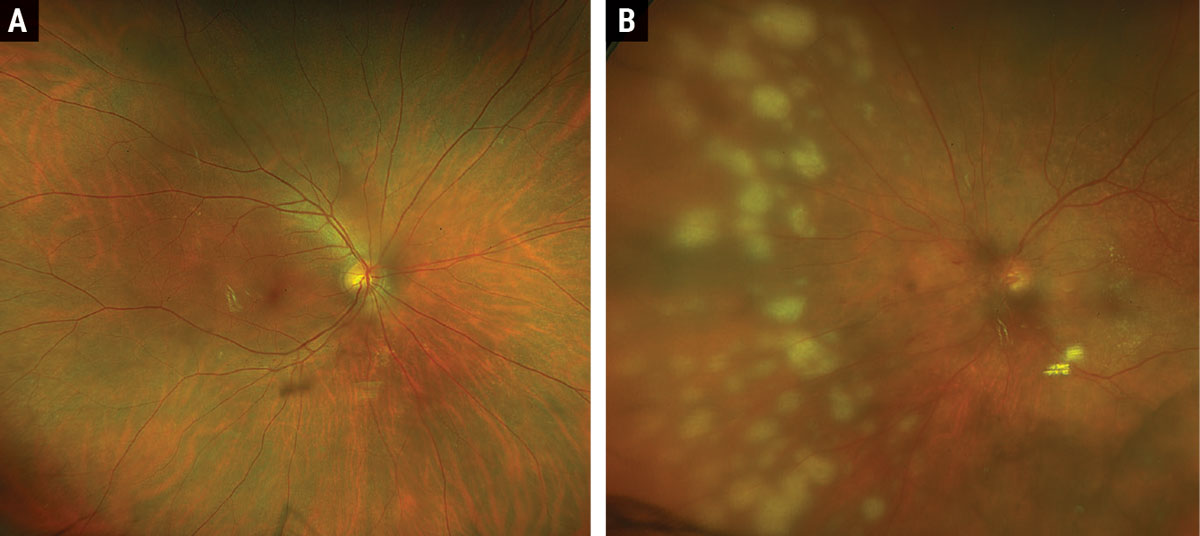 |
Fig. 1. Widefield Optos fundus photos of right (A) and left (B) eyes. Click image to enlarge. |
Diagnosis
Fundus exam OD showed a posterior vitreous detachment and few intraretinal hemorrhages along the inferotemporal arcade (Figure 1). Fundus examination OS disclosed sheets of vitreous cells, peripapillary intraretinal hemorrhages, peripapillary and macular exudates and numerous round, creamy-white retinal infiltrates dispersed throughout the nasal equatorial fundus (Figure 1). Fluorescein angiography showed early hypofluorescence and late hyperfluorescence of the lesions (Figure 2). The patient underwent a diagnostic pars plana vitrectomy and fine needle aspiration biopsy of the lesions. Immunohistochemistry showed atypical CD20 positive lymphoid cells, and cytogenetics showed that the MYD88 gene contained an L265P point mutation, consistent with a diagnosis of primary vitreoretinal lymphoma.
Discussion
Primary vitreoretinal lymphoma (PVRL) is a rare malignancy that represents the most common manifestation of intraocular lymphoma, followed by uveal lymphoma.1,2 PVRL is primarily a high-grade lymphoma and a subset of primary central nervous system lymphoma (PCNSL) that occurs when the retina and vitreous are the primary site of involvement (about 20% of patients).1,2 Importantly, up to 25% of patients presenting with PCNSL will also show symptoms of PVRL or go on to development it, and up to 90% of patients with PVRL are expected to have central nervous system (CNS) disease or to progress to CNS involvement within 29 months, which is the typical etiology for associated disease mortality.1,2
Epidemiology
Most patients present later in life, typically in the fifth and sixth decades, with no definitive gender predilection.1,3 Given its rarity, exact incidence of PVRL is unknown, but estimated to be between 50 and 380 annual cases in the United States, and PCNSL is estimated to be about 1,900 annual cases in the US.2,4 Presenting VA can range from 20/20 to hand motions and median overall survival time is approximately 58 months, neither of which are dependent upon local interventional approach.1,2 The only identified risk factors for development of PVRL seem to be immunodeficiency (e.g., human immunodeficiency virus; HIV) and Epstein-Barr virus infection.3
Pathophysiology
Our understanding of PVRL supports that clonal proliferation of malignant lymphocytes likely occurs outside the CNS, within the systemic circulation.1 Subsequently, these malignant lymphocytes infiltrate the eye and brain, presumably reaching the eye via retinal endothelial receptors; immunocompromise and reduced local immunosurveillance may permit or facilitate cellular infiltration.1
In PVRL, the vitreous humor, neurosensory retina, RPE and optic nerve may be infiltrated, with Bruch’s membrane serving as a barrier for further dissemination into the choroid/uveal tract, which is notably not involved in this disease.1,4 In contrast, metastatic systemic lymphoma to the eye involves the uveal tract, most commonly via the rich choroidal circulation.1 Additionally, primary uveal lymphoma, also referred to as benign reactive lymphoid hyperplasia, is felt to be a low-grade B-cell lymphoma with little/no metastatic potential, though can still cause local organ morbidity.1
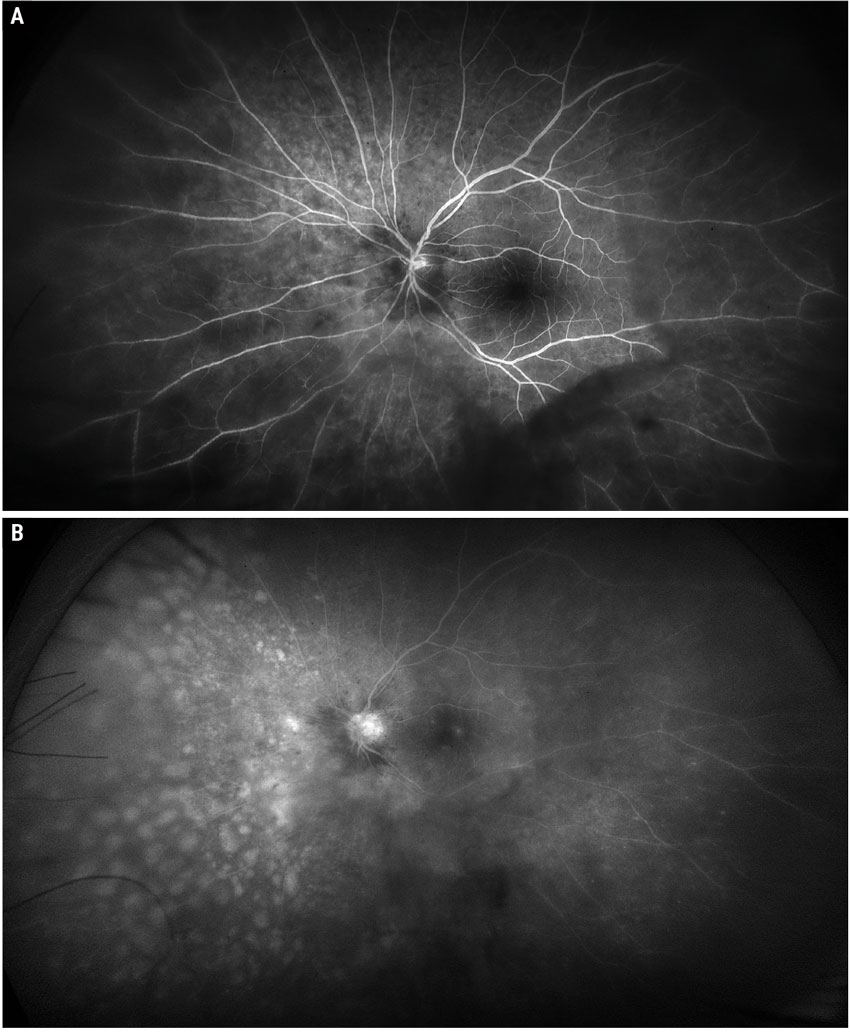 |
Fig. 2. Widefield Optos fluorescein angiogram early and late phase of the left eye. Click image to enlarge. |
Differential Diagnosis
All the intraocular lymphomas are uveitis masquerade syndromes. First used in 1967 to describe a chronic conjunctivitis secondary to carcinoma, the term “masquerade syndrome” has since been adapted to refer to any disease in which there is cellular infiltration mimicking the appearance of intraocular inflammation in the absence of a truly immune-mediated or infectious process.5
Differential diagnoses for primary vitreoretinal lymphoma primarily include different infectious and inflammatory etiologies of intermediate and posterior uveitis such as syphilis, tuberculosis, toxoplasmosis, viral retinitis, sarcoidosis, birdshot chorioretinitis and infectious endophthalmitis.
Clinical and Multimodal Imaging Features
Typical clinical features of PVRL include a moderate, non-clumped cellular reaction in the vitreous cavity, with cells often arranged in sheets, and white/yellow/orange retinal infiltration.1,2 Malignant lymphoid cells often begin along Bruch’s membrane and proliferate in the sub-RPE space, with subsequent RPE alterations and migratory clumping overlying the infiltrate; this may produce the typical leopard-spotted appearance.2,6 Presence of symptomatic iritis (i.e., redness, pain, photophobia) and posterior synechiae should prompt reconsideration of diagnosis.
FAF imaging may show normal, hypo- or hyperAF based on depth and extent of lesions present, as well as overlying lipofuscin and RPE changes.1,2,6 Fluorescein angiography (FA) generally shows hypofluorescence of the lesions in the early and late phases of the angiogram but can also be hyperfluorescent as well.2,4 Rarely, there may be retinal venous leakage, periarteriolar staining and cystoid macular edema.1,2 OCT demonstrates hyperreflective nodular deposits at the level of the RPE, and there may be confluence and elevation in the subretinal or sub-RPE space over time.2,6 While FAF may be normal, a “granular” pattern on FAF is highly predictive of PVRL, the hyperAF lesions correlate with hypofluorescence on FA in about half of the cases and hyperreflective nodularity of the RPE on OCT in about one third of cases.6
Diagnosis
Making the diagnosis first requires a high degree of clinical suspicion to initiate the proper workup. Patients with chronic noninfectious posterior or panuveitis not responding to anti-inflammatory therapy should raise suspicion for a masquerade syndrome, e.g., PVRL.
Anterior chamber paracentesis testing of aqueous humor for elevated levels of interleukin (IL)-10, as well as elevated ratio of IL-10 to IL-6, have been proposed as a screening modality as it is indicative of possible PVRL.1,2,4 IL-10 serves as a scaffold for B-cell lymphocyte proliferation and is also anti-inflammatory, resulting in the typical “white and quiet” appearing eye without an overt inflammatory reaction in response to uncontrolled cellular proliferation.1 Immune-mediated uveitis generally has elevated IL-6, which is associated with breakdown of the vitreous humor structure, and there may be subsequent stranding and aggregations of vitreous opacities.1
Definitive diagnosis requires identification of malignant intraocular lymphocytes in vitreous humor specimen obtained either via direct vitreous aspiration or pars plana vitrectomy.1,3 Since lymphoid cells are fragile, the cytology sample must be analyzed promptly to avoid necrosis and false negative studies.3 Typical immunohistochemistry stains employed to detect most B-cell lymphomas include CD20 and kappa/lambda light chain markers, though these are limited in detecting rarer, more aggressive, T-cell lymphomas.1 Cytogenetics have identified mutations in MYD88 and CD79B as the two most prevalent mutations, present in 88% and 35% of patients with PVRL, respectively.3,4
Management and Prognosis
When presenting as isolated PVRL without CNS involvement, local therapy is recommended and options include local radiation and intravitreal chemotherapy with methotrexate or rituximab.1,2 When presenting with concomitant ophthalmic and CNS disease, high-dose systemic methotrexate with or without rituximab is combined with local therapy.2,3
The prognosis is generally poor, with mortality rates ranging from 9% to 81% and a five-year overall survival rate of less than 25%.3 Treatment is effective at local tumor control but does not alter CNS disease or overall survival.3 Vision is generally preserved until there is direct cellular infiltration of the central macula. The alterations to the retina and RPE, once damaged, are irreversible even once necessary therapy has been initiated.6
Our patient received 10 intravitreal injections of methotrexate OS administered approximately every 10 days. Vision improved one line to 20/100, though she eventually developed vitreous cells OD for which she also received 10 injections of methotrexate. She has been followed off treatment for six months OD and 18 months OS without intraocular recurrence OU or CNS disease development on serial neuroimaging surveillance.
Retina Quiz Answers
1: d, 2: c, 3: a, 4: d, 5: d
Dr. Aboumourad currently practices at Bascom Palmer Eye Institute in Miami. He has no financial disclosures.
1. Davis JL. Intraocular lymphoma: a clinical perspective. Eye 2013;27(2):153-62. 2. Chan CC, Rubenstein JL, Coupland SE, et al. Primary vitreoretinal lymphoma: a report from an International Primary Central Nervous System Lymphoma Collaborative Group symposium. Oncologist 2011;16(11):1589-99. 3. Kalogeropoulos D, Vartholomatos G, Mitra A, et al. Primary vitreoretinal lymphoma. Saudi J Ophthalmol. 2019;33(1):66-80. 4. Soussain C, Malaise D, Cassoux N. Primary vitreoretinal lymphoma: a diagnostic and management challenge. Blood 2021;138(17):1519-34. 5. Theodore FH. Conjunctival carcinoma masquerading as chronic conjunctivitis. Eye Ear Nose Throat Mon. 1967;46(11):1419-20. 6. Ishida T, Ohno-Matsui K, Kaneko Y, et al. Fundus autofluorescence patterns in eyes with primary intraocular lymphoma. Retina 2010;30(1):23-32. |

